Risk management file structure made easy for ISO 14971 compliance
Jan 23, 2023
ISO 14971 requires manufacturers to establish and maintain a risk management file for their medical devices. A risk management file (RMF) is an essential part of the risk management system, but it proves to be challenging in practice to establish and efficiently maintain risk management files across a diverse portfolio.
Most manufacturers struggle to demonstrate traceability and completeness in their RMF
According to ISO 14971:2019, documentation in the RMF must provide traceability of risk management activities for each identified hazard. It should also provide sufficient evidence of completeness of appropriate treatment of all known and potential risks.
Sounds simple, but difficult to satisfy these expectations in practice.
This is primarily because of the following reasons:
- Limited understanding of the documentation requirements and expectations for the RMF
- Lack of organization and connectivity in documentation reflecting the output of risk management activities
- Challenges in maintaining the RMF through required updates during the device lifecycle
Let us first understand the ISO 14971 requirements and expectations
Traceability: ISO 14971:2019 requires traceability for each identified hazard to the risk analysis, risk evaluation, implementation and verification of risk control measures and the evaluation of the residual risk (Clause 4.5).
Completeness: ISO 14971:2019 requires that risks from all identified hazardous situations have been considered and all risk control activities are completed (Clause 7.6).
As an example, if you have identified 5 potential hazards, each associated with 5 different hazardous situations, where each hazardous situation could lead to 5 different harms, you would have at least 125 individual risk items in your risk analysis (5x5x5=125).
It is clear from this simple example that the number of hazard-harm combinations can quickly get out of control. There are many different techniques that can be used to eliminate irrelevant combinations, but it is also very important to build a documentation structure that allows us to organize outputs from different types of risk analysis in a simple, connected manner.
It is not uncommon for manufacturers to struggle in an audit when they try to demonstrate traceability and completeness using one or more Failure Mode Effect Analysis (FMEA) records.
Here is a simple documentation structure you can use in your RMF
If you are looking for a medical device risk management file example, this simple documentation structure can help you.
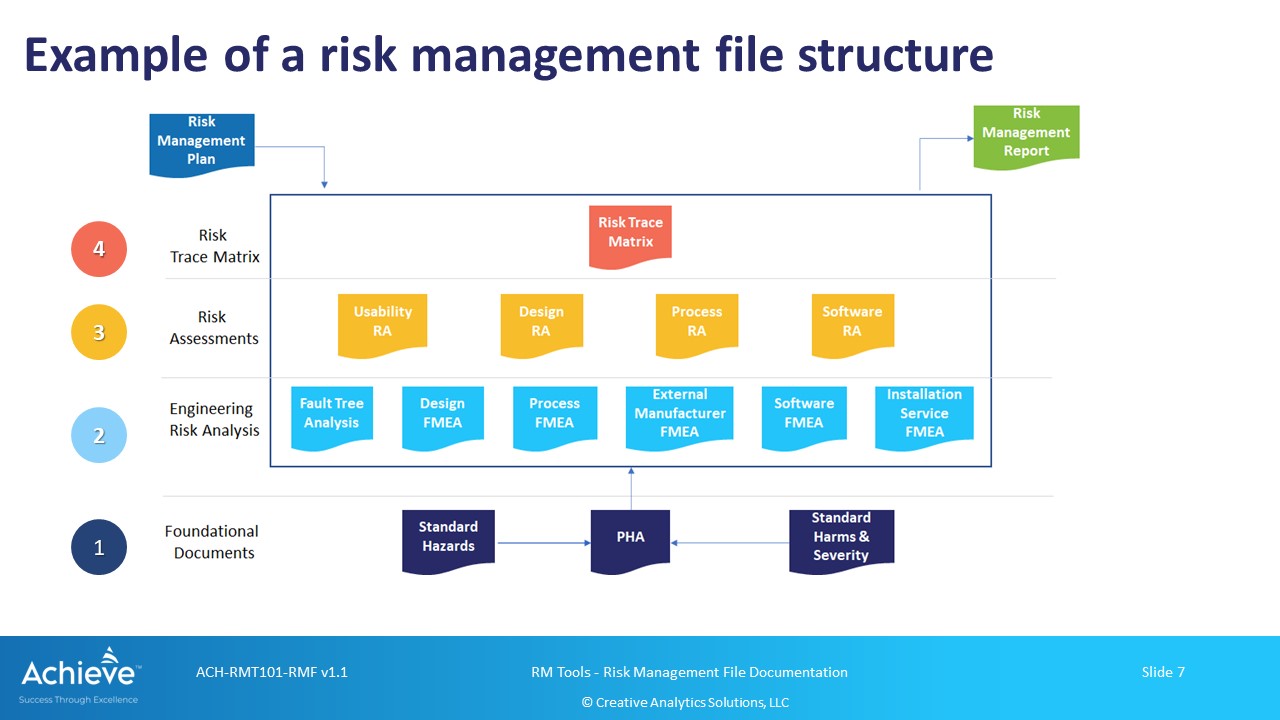
As shown in the figure above, the Risk Trace Matrix is the single record that demonstrates both traceability and completeness of the risk management process in the context of a specific risk management plan (RMP).
The other two key components of the RMF are the risk management plan (RMP) and the risk management report (RMF). Records related to specific risk management activities are shown at levels 2 and 3, which should also be included in the RMF.
If you organize your documentation structure in this way, you will find compliance to ISO 14971 requirements will be very easy!
Level 1: Foundational Records
A standards hazard table, a preliminary hazard analysis (PHA) and a standard harms table are the three foundational records that can be used as a starting point for a portfolio of diverse medical devices. They are not explicitly included in the RMF for a specific medical device (or family) but may be referenced in the RMP.
Level 2: Failure Analysis Records
At level 2, you may have records from different types of failure analysis. As an example, you may have dFMEA (design), pFMEA (process), sFMEA (software), uFMEA (use-misuse) etc. You may also have one or more outputs from a Fault Tree Analysis (FTA).
However, it is very important to note that these records represent analysis of potential failures, and appropriate mitigating controls. They do not represent risk analysis in the context of ISO 14971 because you are not analyzing the risk of harm in these activities.
Certainly, potential failure modes, or combinations thereof, may be related to one or more hazard-harm combinations, but not always. Therefore, wherever appropriate, you can indicate a link to hazard-harm in these records. This linkage is useful to then aggregate all risk control measures for each hazard-harm combination in the risk assessment records (Level 3).
Level 3: Risk Assessment Records
At level 3, you are now compiling information from underlying failure analysis activities in the context of specific hazard-harm combinations reflected in your hazard analysis.
Remember, risk assessment involves not only risk analysis (identification + estimation), but also risk evaluation base on criteria defined in the RMP. It also shows the residual risk level for each individual risk, and benefit-risk analysis if applicable.
At this level, you can have a Design Risk Assessment (DRA) mapped to a dFMEA, a Process Risk Assessment (PRA) mapped to a pFMEA, a Software Risk Assessment (SRA) mapped to a sFMEA, and a Use-Error Risk Assessment (URA) mapped to a uFMEA.
This separation is required because each hazard-harm may be linked to one or more potential failure modes in the underlying mapping record.
Level 4: Risk Trace Matrix
At the highest level 4, you are now aggregating the outputs from the risk assessments at level 3. This is the single, overall record that represents the cumulative output from your risk management activities.
It may appear highly burdensome, and possibly redundant, to establish and maintain this documentation structure in a manual process that mainly utilizes Excel worksheets. However, this process can be easily automated and streamlined using commercially available relational database software.
Even in a manual operation, we have successfully deployed this documentation structure for multi-billion dollar medical devices supported by a small risk management team.
Maintain your RMF throughout the device lifecycle with a simple checklist and a Quality Plan
Another area of practical difficulty is to maintain multiple RMFs throughout the product lifecycle.
Medical devices generally go through a lot of changes during their lifecycle in response to the experience gained during the post-market phase. It is not uncommon to have multiple cycles of design changes to fix newly discovered hazards, hazardous situations and/or device malfunctions.
Knowing exactly which record is included in your RMF, and for what purpose, is very important. You can download our free risk management file checklist here, that also provides you detailed guidance on each type of document or record.
Finally, establishing a Quality Plan is very useful to manage changes to documents and records. Remember that your RMF must be current and complete at all times. Therefore, all documents and records in the RMF must be updated in a timely way.
This can prove to be very burdensome! Therefore, it is best to establish a risk-based approach to updating RMF documents through a Quality Plan. As an example, activities related to newly identified hazards, hazardous situations, harms and safety-critical device failures should be given a higher priority. Minor editorial changes and/or format changes may be done in bulk, at a lower frequency. However, it is very important for you to clearly outline your risk-based approach and document update frequency in the Quality Plan.
In Conclusion:
Establishing and maintaining a risk management file (RMF) is not an easy task. It becomes especially burdensome when you have large portfolio of diverse medical products. Here are a few tips and best practices:
- Build a solid understanding of ISO 14971 requirements.
- Establish a simple and connected documentation structure
- Establish a Quality Plan with a risk-based approach to updating documents in your RMF
Check out our Easy Risk Management File Structure mini-course for more guidance and resources! It includes a checklist and step-by-step directions to help you comply with ISO 14971 requirements.
As a bonus, you will also get tips on audit preparation. Click the image below to access this mini-course.
